Pasqualoto K. F. M., Ferreira M. M. C., "Molecular modeling and QSAR studies of a set of diazaborines - an experimental class of antibacterial agents". Porto de Galinhas, PE, Brazil, 09-13/11/2008: The 4th Brazilian Symposium on Medicinal Chemistry (BrazMedChem2008): Systems Chemical Biology, CD-ROM Online, (2008) No. 152. Poster R0319-1.
Brazilian Chemical Society (SBQ). Division of Medicinal Chemistry. 4th Brazilian Symposium on Medicinal Chemistry
Molecular modeling and QSAR studies of a set of diazaborines - an experimental class of antibacterial agents
Pasqualoto, K. F. M. and Ferreira, M. M. C. - kerlyfmp@iqm.unicamp.br
Laboratory for Theoretical and Applied Chemometrics, Institute of Chemistry, UNICAMP, Campinas - SP, Brasil
Keywords: Molecular Modeling,
QSAR, Diazaborines, Ligand-Based Design, Enoyl-ACP Reductase.
Introduction
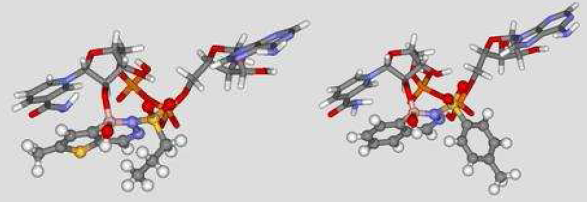
Diazaborines (DBs) represent a group of antibacterial agents of which the important structural element is a heterocyclic 1,2-diazine ring containing a boron as a third hetero atom. The arene group can be benzene, naphthalene, thiophene, furan and pyrrole. The antibacterial activity is confined almost exclusively to Gram-negative bacteria.1,2 Recently, the molecular target of DBs was identified as an enoyl-acp reductase (ENR). ENR catalyzes the last reductive step in the cyclic process of fatty acid elongation, and it is considered the key enzyme of the bacterial fatty acid synthase (FAS II) pathway. The presence of the cofactor nicotinamide adenine dinucleotide (NAD) is required for both the inhibition and the binding of DBs to the ENR enzyme.2 The analysis of the X-ray crystallographic structures of ENR-NAD-DBs complexes revealed the formation of a covalent bond between the 2-hydroxyl of the nicotinamide ribose and the boron atom of the ligands to generate a tight, noncovalently bound bisubstrate analogue (Fig. 1).3 In this study, molecular modeling and QSAR methodologies were applied to a set of fifty-one DB derivatives aiming the rational design of new antibacterial/antimycobacterial agents.
Figure 1. Three dimensional
adduct models, thieno-DB/NAD and benzo-DB/NAD, from 1dfh and 1dfg PDB complexes,
respectively.
Results and Discussion
A set of fifty-one DB derivatives
were selected from ref. [1]. Biological activities were evaluated as the
minimum inhibitory concentration, MIC (mg/mL),
against E. coli D120
at 310 K. These data were converted to molar units and then expressed in
negative logarithmic units, pMIC (-log MIC). The range of activity for
the analogues is more than 3 pMIC units (3.22 to 5.87). The 3D models of
each DB derivative in their neutral forms were constructed using the HyperChem
7.5 software. The crystallized structures of the thieno-DB/NAD and benzo-DB/NAD
adducts (Fig. 1) were used as reference geometries in the building up of
the investigated ligands. Each model was energy-minimized using MM+ force
field without any restriction (HyperChem 7.5). Partial atomic charges were
computed employing the AM1 semiempirical method. The MOLSIM 3.2 program
was also used to optimize the ligands geometry. The molecular dynamics
(MD) simulations protocol included 100,000 steps with a step size of 1
fs at 310 K (T of the biological assay) (MOLSIM 3.2). An output trajectory
file was saved every 20 simulation steps, resulting in 5,000 conformations.
The lowest energy conformation of each ligand was selected and its solvation
and hydrogen bonding energy contributions were calculated. Electrostatic
potential partial atomic charges (Chelpg) were obtained using the ab initio
method HF/6-31G* (Gaussian 03). Thermodynamic descriptors from MD simulations
and other calculated descriptors (electronic, lipophilic, steric and structural),
totalizing 28 independent variables, were considered in the construction
of QSAR models. Partial least squares (PLS) regression and genetic function
approximation (GFA),4
implemented in WOLF 5.5 program, were used as fitting functions. Preliminary
analysis detected one outlier in the investigated set. The best QSAR model
(training set, N = 39; q2 = 0.59;
r2
= 0.86; LSE = 0.11; and LOF = 0.16) was validated employing the leave-multi-out
and y-randomization methods. Its external prediction power (test
set, N = 11) was 90.9%.
Conclusions
Thermodynamic descriptors,
molecular volume and the electrostatic charge of sulfur atom in DBs organossulfonyl
side chain present important contributions for the antibacterial activity.
These findings must be considered for designing new antibacterial/antimycobacterial
agents.
Acknowledgements
The authors are grateful
to FAPESP for the financial support.
1 Grassberger, M. A.; Turnowsky,
F.; Hildebrandt, J., J. Med. Chem. 1984, 27, 947;
2 Pasqualoto, K.F.M.; Ferreira,
M.M.C., QSAR Comb. Sci. 2006, 25, 629;
3 Baldock, C.; de Boer, G.
J.; Rafferty, J. B.; Stuitje A.R.; Rice, D.W., Biochem. Pharmacol. 1998,
55,
1541;
4 Rogers, D.; Hopfinger, A.J.,
J.
Chem. Inform. Comput. Sci. 1994, 34, 854.
4th Brazilian Symposium on Medicinal Chemistry - BrazMedChem2008