Vazquez P. A., Barbosa E. G., Ferreira M. M. C., "Desempenho do método TD-M052X no cálculo de energias verticais de excitação eletrônica coparado a outros métodos TD-DFT, CCSD e CASPT2" ["Effectiveness of the TD-M052X method for the calculation of vertical electronic exciation energies compared to other methods TD-DFT, CCSD and CASPT2"]. Fortaleza, CE, 30/05-02/06/2009: 32a Reunião Anual da Sociedade Brasileira de Química - Para Uma Potência Emergente [32nd Annual Meeting of the Brazilian Chemical Society - For An Emergent Potency], CDROM online, (2009) T0457-2. Poster QT-011.
Sociedade Brasileira de Química (SBQ)
Desempenho do método
TD-M052X no cálculo de energias verticais de excitação
eletrônica comparado a outros métodos TD-DFT, CCSD e CASPT2.
Pedro Antonio Muniz Vazquez
(PQ), Euzébio G. Barbosa* (PG), Márcia Miguel Castro Ferreira
(PQ)
euzebiob@iqm.unicamp.br
Instituto de Química, UNICAMP, São Paulo-SP.
Palavras Chave: Excitações eletrônicas,teoria do funcional da densidade, CCSD, CAS PT2.
Introdução
O novo metafuncional híbrido
de troca e correlação M05-2X1
melhorou drasticamente, em relação aos outros funcionais,
o desempenho do tratamento de interações não-covalentes.2
Tais melhorias permitem a inserção da teoria do funcional
de densidade no estudo de sistemas onde as interações não
ligadas devem ser corretamente representadas. M05-2X, implementado recentemente
no programa Gaussian03 (versão E01), mostra-se capaz de representar
diversas propriedades de moléculas orgânicas com exatidão
superior a outros métodos funcionais.3
Devido às diversas
melhorias deste funcional resolvemos investigar o seu desempenho no cálculo
de excitações eletrônicas (TD-DFT) e compara-lo com
outros métodos DFT e CASPT2 usando o método CCSD como referência.
Resultados e Discussão
Oito estados eletrônicos
da geometria cristalográfica da molécula de imidazol4
foram investigados usando os funcionais M05-2X, B3LYP, B3LYPCS005,
PBE0 e os resultados comparados com métodos de ordem superior perturbativos
multiconfiguracional CASPT2 e o método Coupled Cluster com excitações
simples e duplas. A função de base aug-cc-pVDZ foi usada
em todos os métodos citados. Os resultados são mostrados
na Tabela 1.
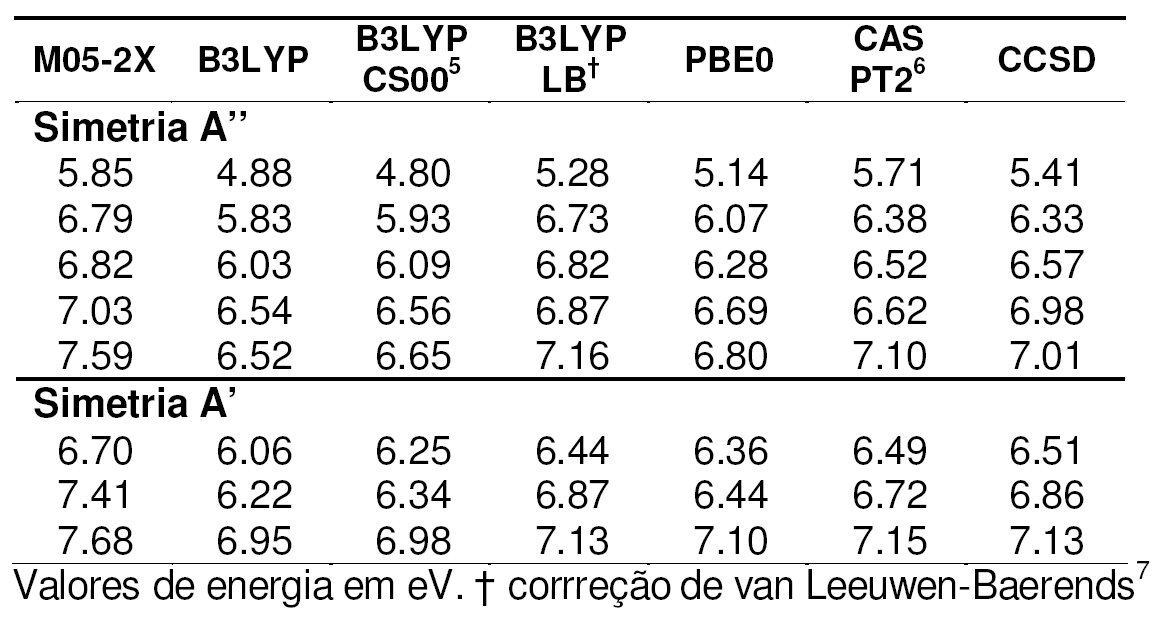
Tabela 1. Energias
dos estados excitados para o imidazol.*
O desempenho dos métodos pode ser melhor analisado com o auxílio da Tabela 2 que mostra os desvios relativos entre os valores calculados para os métodos DFT e CASPT2 em relação a CCSD.
Tabela 2. Desvios
relativos dos métodos DFT e CASPT2
em relação
a CCSD no cálculo de estados excitados do imidazol.*
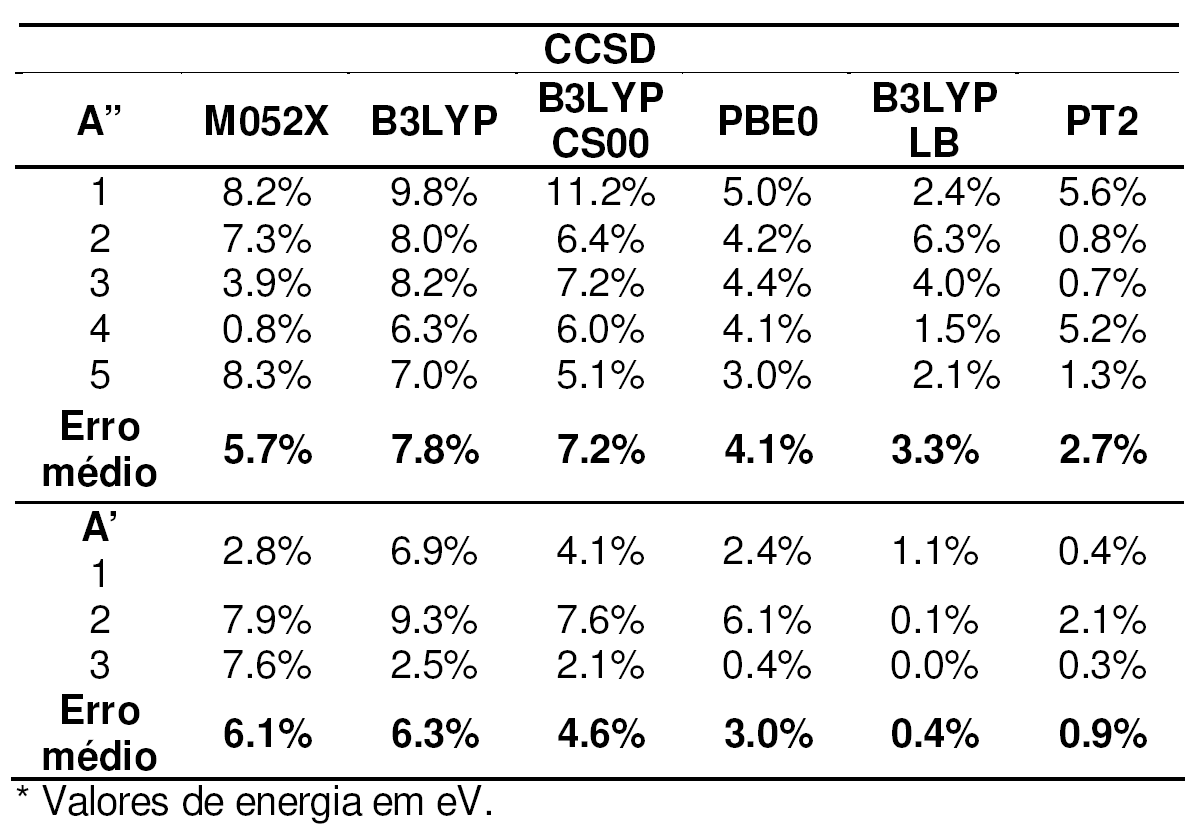
Os resultados mostram que o funcional PBE0 e a correção de Leeuwen-Baerends7, otimizados para o estudo de energias de excitações, se destacaram em relação aos outros funcionais enquanto o M05-2X teve desempenho aquém às expectativas.
Conclusões
Dos resultados obtidos conclui-se
que, para esta molécula, o funcional M05-2X, embora uma potencial
alternativa ao popular B3LYP, não é vantajoso para o cálculo
de energias de excitações apresentando performance inferior
ao PBE0. O seu desempenho na otimização de geometria de estados
excitados do imidazol está sendo investigado.
Agradecimentos
Os autores gostariam de
agradecer a CAPES e CNPQ pela ajuda financeira.
____________________________
1Zhao, Y., Schultz,
N.E.; Truhlar, D. G. J. Chem. Theory Comput. 2006, 2, 364.
2Zhao, Y; Truhlar,
D. G. J. Chem. Theory Comput. 2007, 3, 289.
3Zhao, Y; Truhlar,
D. G. Org. Lett., 2006, 8, 5753.
4Craven, B. M.
McMullan, R. K. Bell, J. D.; Freeman H. C. Acta Cryst. 1977, B33.
2585.
5Casida M.E.;
Salahub, D.R. J. Chem. Phys. 2000, 113, 8918.
6Serrano-Andrs,
L., Flscher, M. P., Roos, B.O.; Merchn, M. J. Phys. Chem., 1996,
100, 6484.
7van Leeuwen,
R.; Baerends, E.J. Phys. Rev. A. 1994, 49, 2421.
32a Reunião Anual
da Sociedade Brasileira de Química
English
Sociedade Brasileira de Química (SBQ)
Effectiveness of the TD-M052X
method for the calculation of vertical electronic exciation energies compared
to other methods TD-DFT, CCSD and CASPT2.
Pedro Antonio Muniz Vazquez
(PQ), Euzébio G. Barbosa* (PG), Márcia Miguel Castro Ferreira
(PQ)
euzebiob@iqm.unicamp.br
Instituto de Química, UNICAMP, São Paulo-SP.
Key Words: Electronic excitations, density functional theory, CCSD, CAS PT2.
Introduction
The new hybrid exchange
and correlation metafunctional M05-2X1
has been improved significantly in treatment of non-covalent interactions2,
when compared to other functionals. This makes the density functional theory
useful in studies of systems in which the non-bonding interactions have
to be included correctly. M05-2X was recently implemented in program Gaussian03
(version E01), and it has been shown capable to treat diverse properties
of organic molecules with accuracy superior to that of other functionals.3
The mentioned improvements
were the reason to investigate the performance of this functional in calculations
of electronic excitations (TD-DFT) and compare it with other DFT and CASPT2
methods, with the CSD method as the reference.
Results and Discussion
Eight electronic states
of a crystallographic geometry of imidazole molecule4
were studied by using functionals M05-2X, B3LYP, B3LYPCS005
and PBE0. Obtained results were compared to those from higher order perturbation
methods multiconfigurational multiconfiguracional CASPT2 and Coupled Cluster
method for single and double excitations. The basis function aug-cc-pVDZ
was used in all the methods. The results and shown in Table 1.
Table 1.
Energies of excited states of imidazole.*
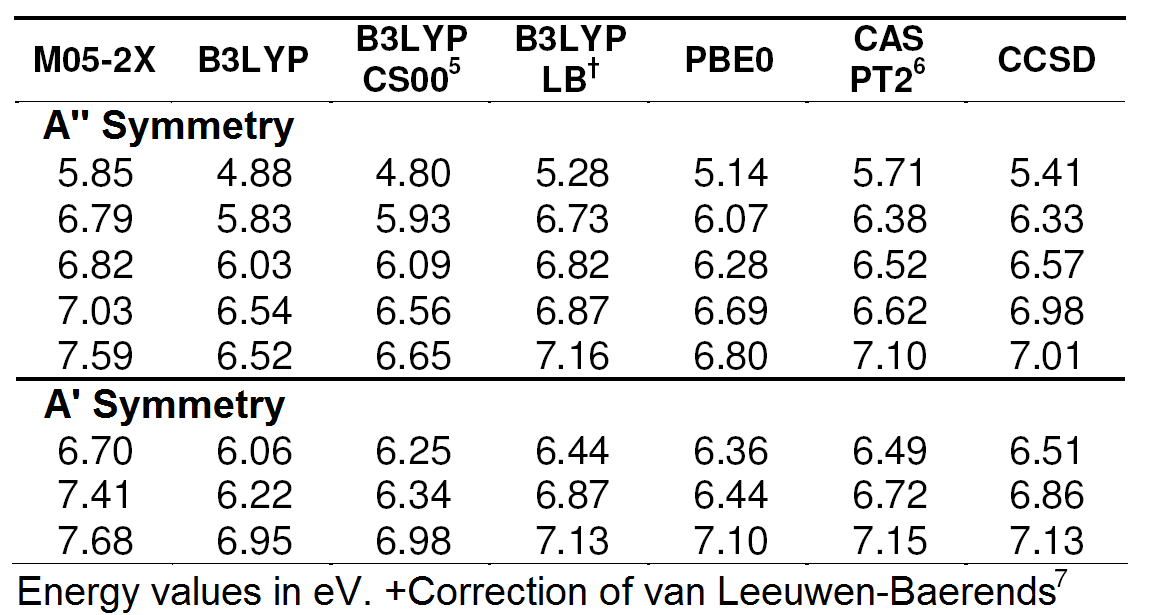
Table 2 can aid in analyzing the performance of the methods used, by which relative errors of the methods DFT and CASPT2 are compared to those of CCSD.
Table 2.
Errors of the methods DFT and CASPT2 relative
to CCSD in calculation of
excited states of imidazole.*
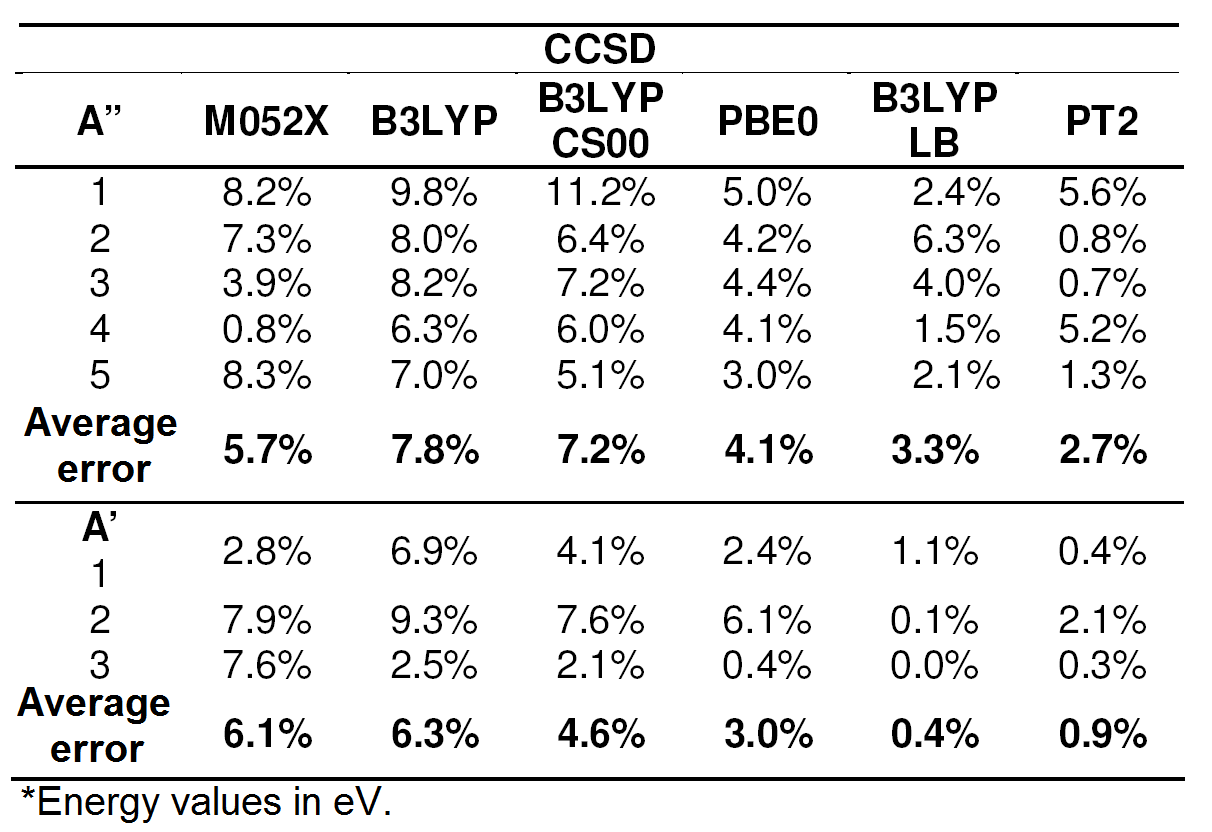
According to these results, the PBE0 functional and the Leeuwen-Baerends correction7 oare outstanding among the all functional used for calculation excitation energies, whilst the M05-2X functional has performance beyong expectations.
Conclusions
It can be concluded for
the studied molecule that the M05-2X functional, although being a potential
alternative to the popular B3LYP, is not preferred for excitation
energy calculations due to its inferiority with respect to functional PBE0.
Its performance in geometry optimization of excited states of imidazole
is being studied.
Acknowledgements
The authors would like to
thank CAPES and CNPQ for financial aid.
____________________________
1Zhao, Y., Schultz,
N.E.; Truhlar, D. G. J. Chem. Theory Comput. 2006, 2, 364.
2Zhao, Y; Truhlar,
D. G. J. Chem. Theory Comput. 2007, 3, 289.
3Zhao, Y; Truhlar,
D. G. Org. Lett., 2006, 8, 5753.
4Craven, B. M.
McMullan, R. K. Bell, J. D.; Freeman H. C. Acta Cryst. 1977, B33.
2585.
5Casida M.E.;
Salahub, D.R. J. Chem. Phys. 2000, 113, 8918.
6Serrano-Andrs,
L., Flscher, M. P., Roos, B.O.; Merchn, M. J. Phys. Chem., 1996,
100, 6484.
7van Leeuwen,
R.; Baerends, E.J. Phys. Rev. A. 1994, 49, 2421.
32a Reunião Anual
da Sociedade Brasileira de Química