Pereira F. S., Pasqualoto K. F. M., Ferreira M. M. C., "QSAR MULTIVARIADO DE UM CONJUNTO DE COMPOSTOS COM ATIVIDADE ANTIMALÁRICA" ["MULTIVARIATE QSAR FOR A SET OF COMPOUNDS WITH ANTIMALARIAL ACTIVITY"]. Poços de Caldas, MG, 18-21/11/2007: XIV Simpósio Brasileiro de Química Teórica [14th Brazilian Symposium of Theoretical Chemistry], Resumos [Abstracts], (2007) 193. Poster.
Sociedade Brasileira de Química
(SBQ)
QSAR MULTIVARIADO DE UM CONJUNTO DE COMPOSTOS COM ATIVIDADE ANTIMALÁRICA
Flávia da Silva Pereira (PG), Kerly Fernanda Mesquita Pasqualoto (PQ), Márcia Miguel Castro*. marcia@iqm.unicamp.br
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas SP
Palavras Chave: Malária, Artemisinina, QSAR, PLS
Introdução
Segundo a Organização
Mundial de Saúde (OMS), a malária tornou-se um problema de
saúde publica em 90 países, nos quais cerca de 2,4 bilhões
de pessoas, ou seja, cerca de 40% da população mundial convivem
com os riscos de contágio.1 A OMS
determinou prioritário o desenvolvimento de esquizonticidas sanguíneos
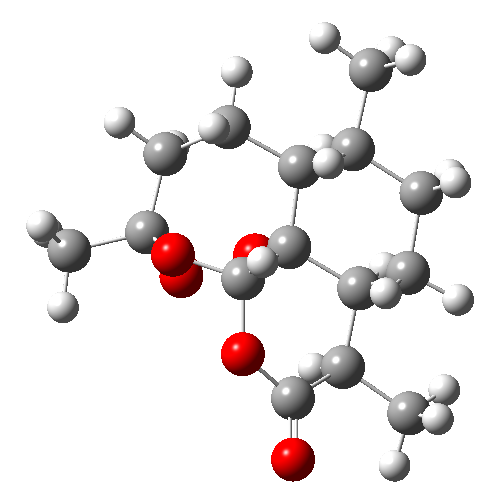
de ação rápida, derivados da artemisinina (Fiqura1),
ao tratamento da forma grave de malária e, também, ao controle
de cepas multirresistentes de P. falciparum.
Figura 1. Estrutura molecular da artemisinina.
Estudos de QSAR (Quantitative Structure Activity Relationships) relacionam estrutura e atividade de conjuntos de compostos, considerando descritores eletrônicos, hidrofóbicos e estéricos. O presente estudo tem como finalidade aplicar a metodologia QSAR a um conjunto de derivados da artemisinina. O fármaco de referência também foi incluído na análise.
Resultados e Discussão
Estudo preliminar foi realizado
com 30 compostos, ensaiados sob um mesmo protocolo farmacológico.
Os compostos foram desenhados considerando como referência as coordenadas
da estrutura da artemisina (CSD versão 1.9). Os métodos empregados
na otimização da geometria dos compostos foram mecânica
molecular (MM+), semi-empírico (AM1) e método Hartree-Fock,
com os conjuntos de bases: HF/3-21G e HF/6-31G.
Os dados de atividade dos
análogos foram obtidos utilizando a artemisinina como referência,2
a partir da seguinte relação:
Log Atividade Relativa (LogRA)
= 100 log[(IC50ARTEMISININA/IC50ANALOGO)X(MMANALOGO/MMAARTEMISININA)]
Descritores estruturais
(distância de ligação, ângulos de ligação
e diedros), eletrônicos (HOMO, LUMO, cargas, polarizabilidade entre
outros) e topológicos (Balabam, Randic entre outros) foram calculados
e utilizados como variáveis independentes na construção
do modelo. O melhor modelo, construído com o conjunto de treinamento
(N=24), apresentou três variáveis latentes com 57,8% de informação
total do sistema, valor de SEVC = 0,44, Rval = 0,65 e Rcal = 0,81. O modelo
apresentou 3 amostras atípicas. O conjunto de validação
externa (N=3) apresentou um erro de previsão satisfatório,
semelhante ao do modelo.
A seleção
dos descritores foi realizada com base no coeficiente de regressão
(programa PIROUETTE)3 e no significado
físico de tais variáveis em relação à
atividade biológica. Os descritores mais representativos ao modelo
estão apresentados na equação abaixo:
LogRA = -0,24m
+ 0,61Eel + 0,27 LUMO 0,38 pR
0,35 BALABAN 0,39 nH + 0,26 qO1 0,26 q C9 + 0,30 O3-C10 0,28 D(O1C9O3C10)
Considerando o modelo obtido,
os descritores que contribuem favoravelmente à atividade são:
Eel, LUMO, qO1 e O3-C1, que são descritores eletrônicos e
estruturais (distância). Os demais são desfavoráveis
à atividade do sistema investigado.
Como perspectiva, outros
compostos serão incluídos no conjunto, além de outros
descritores (descritores termodinâmicos), a fim de extrair informações
mais relevantes. Além disso, outra metodologia de otimização
na construção de modelos será empregada (algoritmo
de aproximação da função genética, GFA,
Genetic Function Approximation).4
Conclusões
O estudo preliminar forneceu
modelo robusto, com destaque aos descritores eletrônicos e estruturais.
O modelo final será construído com inclusão de novos
compostos e descritores termodinâmicos. A metodologia de algoritmo
genético (GFA) também será utilizada na otimização
da construção dos modelos.
Agradecimentos
Os autores agradecem ao
CNPq e a FAPESP.
____________________________
1 Krungkrai,
S. R.; DelFraino, B.J.; Smiley, J.A.; Prapunwattana, P.; Mitamura, T.;
Horii, T.; and Krungkrai, J. Biochemistry, 2005, 44, 1643.
2 Lisgarten,
J.N.; Potter, B.S.; Bantuzeko, C.; Palmer, R.A. J. Chem. Cryst.,
1998, 28, 539.
3 PIROUETTE 3.01,
INFORMETRIX, INC.; Woodinville, WA, 2001.
4 Rogers, D.;
Hopfinger, A.J. J. Chem. Inf. Comput. Sci., 1994, 34, 854.
193
English
Sociedade Brasileira de Química
(SBQ)
MULTIVARIATE QSAR FOR A SET OF COMPOUNDS WITH ANTIMALARIAL ACTIVITY
Flávia da Silva Pereira (PG), Kerly Fernanda Mesquita Pasqualoto (PQ), Márcia Miguel Castro*. marcia@iqm.unicamp.br
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas SP
Key Words: Malaria, Artemisinin, QSAR, PLS
Introduction
According to the World Health
Organization (WHO), malaria has become a serious public health problem
in 90 countries, in which 2.4 billion people or about 40% of world population
is under the risk of infection.1 The WHO
has put as the task development of blood esquizoticides with rapid action,
derivatives of arteminin (Figure 1), for treatment of severe malaria and
cases of multiresistant strains of P. falciparum.
Figure 1. Molecular structure of artemisinin.
QSAR (Quantitative Structure Activity Relationships) studies relate structure with activity of a set of compounds, taking into account electronic, hydrophobic and steric descriptors. Aim of the present study is to apply the QSAR methodology for a set of artemisinin derivatives. The referent compound was also included in the analysis.
Results and Discussion
A preliminar study for 30
compounds, used in the same pharmacological assays, has been performed.
The compounds were modeled based on the coordinates of the referent compound
artemisinin (CSD version 1.9). The methods used for geometry optimization
were molecular mechanics (MM+), semi-empirical (AM1) and Hartree-Fock with
basis functions HF/3-21G and HF/6-31G.
The activity data for analogues
were obtained by defining artemisinin as the reference,2
according to the following relation:
Log Relative Activity (LogRA)
= 100 log[(IC50ARTEMISININ/IC50ANALOGUE)X(MMANALOGUE/MMAARTEMISININ)]
Structural descriptors (bond
lengths, bond and dihedral angles), electronic (HOMO, LUMO, charges, polarizabilty,
among others) and topological (Balabam, Randic, among others) descriptors
were calculated and used as independent variables in contruction of models.
The best model, built with the training set (N=24), resulted in three latent
variables with 57.8% of the total information, and with the values SEVC
= 0.44, Rval = 0.65 and Rcal = 0.81. Three outliers were detected. The
external validation set (N=3) had a satisfactory prediction error similar
to that of the model.
Variable selection was based
on regression coefficients (program PIROUETTE)3
and phyisical meaniong of the variables in relation to the biological activity.
The most important descriptors are included in the following equation:
LogRA = -0.24m
+ 0.61Eel + 0.27 LUMO 0.38 pR
0.35 BALABAN 0.39 nH + 0.26 qO1 0.26 q C9 + 0.30 O3-C10 0.28 D(O1C9O3C10)
Considering the obtaind
model, it can be said that the descriptors which favor the biological activity
are: Eel, LUMO, qO1 and O3-C1, which are electronic and structural descriptors
(distance). The other descriptors do not favor the activity of the systems
under study. In future, other compounds will be included in the study,
together with more descriptors (thermodynamic descriptors) in order to
obtain more relavant information. Besides this, another methodology for
construction of the models will be used (an algorithm of Genetic Function
Approximation, GFA).4
Conclusions
This preliminar studied
resulted in a robust model, with the emphasis on electronic and structural
descriptors. The final model will be constructed including new compounds
and thermodynamic descriptors. The genetic algorith (GFA) methodology also
will be used for construction of models.
Acknowledgements
The authors acknowledge
CNPq and FAPESP.
____________________________
1 Krungkrai,
S. R.; DelFraino, B.J.; Smiley, J.A.; Prapunwattana, P.; Mitamura, T.;
Horii, T.; and Krungkrai, J. Biochemistry, 2005, 44, 1643.
2 Lisgarten,
J.N.; Potter, B.S.; Bantuzeko, C.; Palmer, R.A. J. Chem. Cryst.,
1998, 28, 539.
3 PIROUETTE 3.01,
INFORMETRIX, INC.; Woodinville, WA, 2001.
4 Rogers, D.;
Hopfinger, A.J. J. Chem. Inf. Comput. Sci., 1994, 34, 854.
193